The “Fast-Off” Hypothesis Revisited: What Antipsychotic Binding Kinetics Really Teach Us
 Introduction: Why Do Some Antipsychotics Cause More Motor Side Effects?
Introduction: Why Do Some Antipsychotics Cause More Motor Side Effects?
Antipsychotic medicines remain central to the treatment of schizophrenia, mania, psychosis, severe agitation, and several other psychiatric conditions. Yet, one of the long-standing challenges in psychiatry is balancing symptom control with side-effect burden.
Older “typical” antipsychotics such as haloperidol and chlorpromazine are effective dopamine D2 receptor blockers, but they are more likely to produce extrapyramidal symptoms such as stiffness, tremors, restlessness, slowness, dystonia, and long-term movement disorders. Newer “atypical” antipsychotics such as clozapine, quetiapine, olanzapine, risperidone, amisulpride, and others generally have a lower risk of motor side effects, though they may bring other concerns such as weight gain, sedation, metabolic syndrome, prolactin elevation, or QT effects.
For many years, one elegant explanation attempted to simplify this difference: the fast-off hypothesis.
What Was the Original Fast-Off Hypothesis?
The fast-off hypothesis proposed that atypical antipsychotics come off the dopamine D2 receptor much faster than typical antipsychotics. In simple terms, the idea was this:
A typical antipsychotic “sticks” to the D2 receptor for a long time. This produces strong and sustained dopamine blockade, including in the motor circuits of the brain, leading to extrapyramidal symptoms.
An atypical antipsychotic “blocks and releases” the D2 receptor more quickly. This allows enough dopamine signalling to continue in motor circuits, while still helping psychotic symptoms.
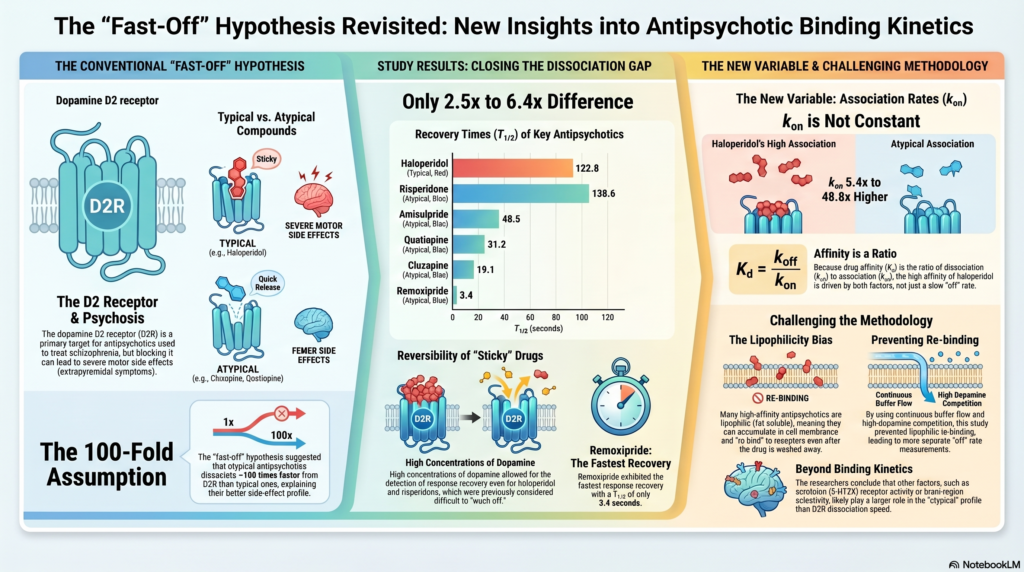
This was a very attractive hypothesis because it explained clinical experience in a neat pharmacological language. The original model suggested that atypical antipsychotics may dissociate from the D2 receptor around 100 times faster than typical antipsychotics. The uploaded paper by Sahlholm and colleagues revisited this assumption using a functional electrophysiological assay of dopamine D2 receptor antagonism.
The Key Pharmacology: k-on, k-off, and Affinity
To understand the debate, we need three terms.
k-on is the association rate: how quickly a drug binds to the receptor.
k-off is the dissociation rate: how quickly the drug leaves the receptor.
Kd, or dissociation constant, reflects affinity. It is not just about how slowly a drug leaves; it is the ratio between leaving and binding:
Kd = k-off / k-on
This is important because an antipsychotic can have high affinity not only because it leaves slowly, but also because it binds quickly. Earlier versions of the fast-off model placed heavy emphasis on k-off and assumed that k-on was relatively similar across antipsychotics. The newer analysis challenges this assumption.
What Did the Revisited Study Find?
Sahlholm and colleagues used a functional assay involving dopamine D2 receptors and GIRK potassium channel responses. Instead of measuring binding only through radioligand displacement, they studied how dopamine-mediated D2 receptor function recovered after antipsychotic antagonism.
The major finding was striking: haloperidol did not appear to be 100 times slower than atypical antipsychotics in terms of functional recovery. In this study, recovery from haloperidol was only about 6.4-fold slower than clozapine, 3.9-fold slower than quetiapine, 2.8-fold slower than olanzapine, and 2.5-fold slower than amisulpride.
This does not mean haloperidol and clozapine are clinically identical. They are clearly different drugs. But it does mean that a simple “typicals are slow-off, atypicals are fast-off” explanation is not enough.
The Problem of “Stickiness”: Is It Receptor Binding or Tissue Retention?
One of the most important insights from the paper is that apparent receptor stickiness may partly reflect lipophilicity.
Lipophilic drugs can accumulate in cell membranes or within cells. If a drug lingers in the membrane, it may appear to remain active at the receptor even after the extracellular drug has been washed away. This can falsely make the drug seem to have a very slow k-off.
Haloperidol is relatively lipophilic. The study suggests that some earlier estimates of very slow dissociation may have been influenced by membrane accumulation and re-binding. In other words, the drug may not necessarily be sitting on the receptor for an extremely long time; it may be lingering nearby and repeatedly re-engaging the receptor.
This distinction matters clinically. A drug’s effects in the brain depend not only on receptor binding, but also on how it distributes into tissue, how long it stays in the brain, how it is metabolised, and how much receptor occupancy it produces over time.
Association Rate Also Matters
The study also found that k-on was not constant across antipsychotics. This is a crucial update.
Haloperidol had a much higher association rate than clozapine and quetiapine. In the study, the estimated k-on for haloperidol was around 5.4 times higher than clozapine and 48.8 times higher than quetiapine. This means haloperidol’s high D2 affinity is not simply because it leaves slowly; it may also bind rapidly and efficiently.
This is where the older model becomes too simplistic. Affinity is a ratio. A drug can look “strong” at D2 receptors because it binds fast, leaves slowly, accumulates in tissue, or some combination of these.
Does This Disprove the Fast-Off Hypothesis?
Not completely. The fast-off hypothesis remains conceptually useful, especially when thinking about drugs like clozapine and quetiapine, which show relatively transient D2 occupancy and lower extrapyramidal risk at therapeutic doses.
But the revised evidence suggests that fast-off kinetics alone cannot explain atypicality.
Atypical antipsychotic action likely depends on several interacting factors:
D2 receptor occupancy level
Speed and duration of D2 occupancy in vivo
5-HT2A receptor antagonism or inverse agonism
Regional selectivity in the brain
Dose and plasma level
Active metabolites
Intrinsic activity or partial agonism in some drugs
Pharmacokinetics, including half-life and tissue distribution
Patient-specific vulnerability to EPS, akathisia, prolactin elevation, sedation, or metabolic effects
This is why clinical psychopharmacology cannot be reduced to one receptor or one kinetic parameter.
Why This Matters in Real-World Psychiatry
For patients and families, the practical message is simple: antipsychotic selection is not about choosing the “strongest” or “newest” medicine. It is about choosing the medicine whose benefit-risk profile fits the person.
For example, a young patient with first-episode psychosis may be highly sensitive to extrapyramidal symptoms and prolactin-related adverse effects. Another patient may be more vulnerable to weight gain and metabolic syndrome. A patient with severe suicidality or treatment-resistant schizophrenia may benefit from clozapine despite its monitoring requirements. A patient with agitation may need a short-term medication strategy that differs from long-term maintenance treatment.
The D2 receptor remains central to antipsychotic efficacy, but good prescribing requires a broader view: receptors, circuits, metabolism, comorbidities, subjective tolerability, adherence, and the patient’s long-term life goals.
A Better Way to Think About Antipsychotics
Instead of asking only, “How tightly does this drug block dopamine?” we should ask:
How much D2 occupancy does this drug produce at this dose?
How long does that occupancy last?
Does it also act on serotonin, histamine, muscarinic, adrenergic, or glutamate-related systems?
Is the patient developing stiffness, restlessness, emotional dulling, sexual dysfunction, sedation, or weight gain?
Is the patient actually able and willing to continue the medication?
Can we use the lowest effective dose while protecting recovery and functioning?
The future of antipsychotic prescribing will be increasingly personalised. We are moving from broad labels like “typical” and “atypical” toward a more precise understanding of receptor profiles, brain networks, pharmacokinetics, digital monitoring, metabolic risk, and patient-reported outcomes.
Clinical Takeaway
The fast-off hypothesis was an important milestone in psychopharmacology. It helped clinicians and researchers think beyond simple dopamine blockade. However, newer functional kinetic evidence suggests that the difference between typical and atypical antipsychotics is not explained by a dramatic 100-fold difference in D2 dissociation alone.
The brain is more nuanced. Antipsychotic action depends on both how a drug binds, how it leaves, where it acts, how long it occupies receptors, and how the individual patient responds.
Good psychiatry lies in this nuance.
Conclusion
Antipsychotics are not interchangeable dopamine blockers. They are complex neurochemical tools with different binding kinetics, receptor profiles, pharmacokinetics, and clinical personalities. The fast-off hypothesis remains useful, but it should not be treated as the single explanation for atypicality or tolerability.
For patients, the most important message is this: side effects are not something to silently tolerate. If a medicine causes stiffness, restlessness, tremors, sedation, emotional blunting, weight gain, sexual side effects, or menstrual changes, it is worth discussing. There are often ways to adjust the dose, switch medicines, add protective strategies, or rethink the treatment plan.
Reference note: This article is based on Sahlholm et al., The fast-off hypothesis revisited: A functional kinetic study of antipsychotic antagonism of the dopamine D2 receptor, European Neuropsychopharmacology, 2016.
Dr. Srinivas Rajkumar T, MD (AIIMS), DNB, MBA (BITS Pilani)
Consultant Psychiatrist & Neurofeedback Specialist
Mind & Memory Clinic, Apollo Clinic Velachery, Chennai
✉ srinivasaiims@gmail.com 📞 +91-8595155808
Related posts:
- How to Teach & Support a Child with Dyslexia: Home & School Strategies
- 🎮 Loot Boxes and the Gateway Hypothesis: Are We Training the Next Generation to Gamble?
- Vilazodone (Vilano, Vilaz, Vilapride, Vilaplex, Viibryd): The Fast-Acting Antidepressant That Fizzled Out
- Fast-Track ADHD Assessment in Chennai
- Pimavanserin: A Breakthrough Antipsychotic for Parkinson’s Disease Psychosis
- Why Are Antipsychotic Medicines Increasingly Used in Older Adults?