Amyloid Synthesis and Degradation: Why Alzheimer’s Disease Is a Disorder of Failed Clearance
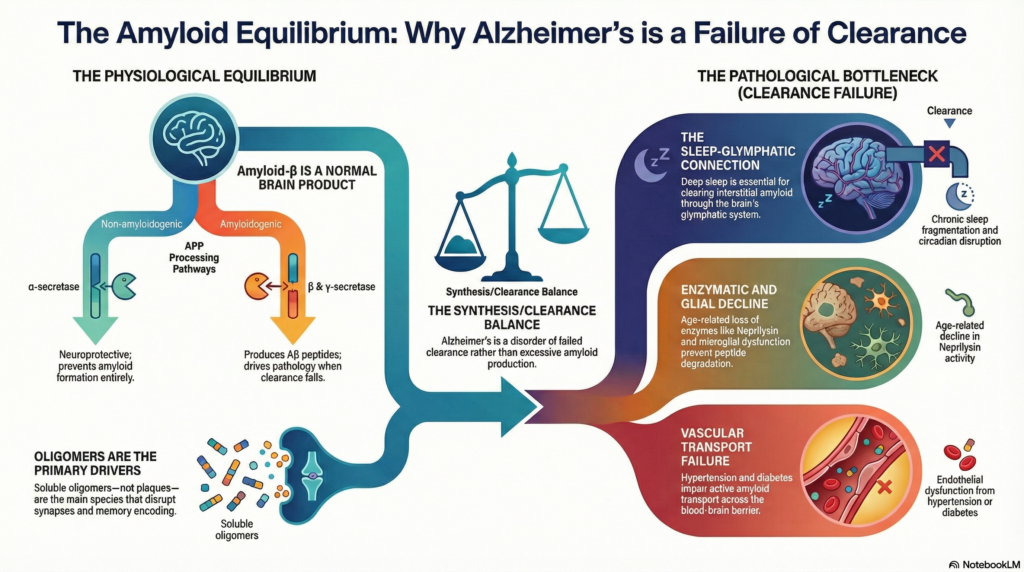 Amyloid-β (Aβ) sits at the center of Alzheimer’s disease biology, yet it is often misunderstood as an abnormal toxin. Contemporary neuroscience tells a more precise story: amyloid is a physiological peptide, produced throughout life, whose pathological significance emerges only when degradation and clearance mechanisms fail. Alzheimer’s disease, in its most common late-onset form, is therefore best understood as a disorder of amyloid homeostasis rather than excessive synthesis.
Amyloid-β (Aβ) sits at the center of Alzheimer’s disease biology, yet it is often misunderstood as an abnormal toxin. Contemporary neuroscience tells a more precise story: amyloid is a physiological peptide, produced throughout life, whose pathological significance emerges only when degradation and clearance mechanisms fail. Alzheimer’s disease, in its most common late-onset form, is therefore best understood as a disorder of amyloid homeostasis rather than excessive synthesis.
Amyloid-β: a normal product of a working brain
Amyloid-β peptides arise from the processing of amyloid precursor protein (APP), a ubiquitous neuronal transmembrane protein involved in synaptic development, plasticity, and axonal transport. APP processing is continuous and tightly regulated; complete suppression is neither biologically realistic nor clinically desirable.
Under physiological conditions, amyloid-β is produced in small quantities and rapidly removed. Pathology arises when this equilibrium is disrupted.
APP processing and amyloid synthesis
APP follows two principal processing pathways.
Non-amyloidogenic pathway (protective)
Cleavage by α-secretase (ADAM10) occurs within the amyloid sequence, producing soluble APPα and preventing amyloid formation entirely. This pathway is promoted by synaptic activity and is associated with neurotrophic and synaptoprotective effects.
Amyloidogenic pathway (constitutive)
Sequential cleavage by β-secretase (BACE1) and γ-secretase generates amyloid-β peptides, predominantly Aβ40 and Aβ42. Aβ42, though less abundant, is more hydrophobic and aggregation-prone, conferring greater neurotoxicity.
Importantly, in sporadic Alzheimer’s disease, there is no convincing evidence of pathological overproduction of amyloid. Instead, subtle shifts in peptide ratios and aggregation kinetics—combined with impaired clearance—drive accumulation over decades.
Oligomers, not plaques, mediate toxicity
Modern evidence consistently implicates soluble amyloid-β oligomers as the principal neurotoxic species. These oligomers:
-
Disrupt glutamatergic receptor trafficking
-
Impair long-term potentiation
-
Alter network oscillations critical for memory encoding
-
Activate tau-phosphorylating kinases
-
Trigger complement-mediated synaptic pruning
Amyloid plaques, while diagnostically useful, represent downstream deposits and correlate poorly with cognitive severity. Their presence reflects chronic failure of amyloid clearance rather than active toxicity.
Amyloid degradation and clearance: the critical bottleneck
The human brain possesses multiple, overlapping amyloid clearance systems.
Enzymatic degradation
The most important amyloid-degrading enzymes include:
-
Neprilysin (NEP) – dominant Aβ42-degrading enzyme
-
Insulin-degrading enzyme (IDE)
-
Endothelin-converting enzymes
Age-related decline in neprilysin activity is one of the most robust biochemical findings in late-onset Alzheimer’s disease.
Glial clearance
Microglia and astrocytes internalize amyloid via phagocytosis. With aging and chronic inflammation, microglia shift to a dysfunctional phenotype, reducing clearance while amplifying neuroinflammation—particularly in individuals carrying the APOE-ε4 allele.
Vascular and BBB-mediated clearance
Amyloid is actively transported out of the brain via LRP1 receptors at the blood–brain barrier. Vascular risk factors—hypertension, diabetes, endothelial dysfunction—impair this mechanism, contributing to cerebral amyloid angiopathy and parenchymal accumulation.
Glymphatic clearance and sleep
During deep slow-wave sleep, convective cerebrospinal fluid flow clears interstitial amyloid. Chronic sleep fragmentation and circadian disruption significantly reduce this clearance, accelerating amyloid accumulation years before cognitive symptoms appear.
Alzheimer’s disease as an amyloid clearance disorder
In late-onset Alzheimer’s disease, amyloid pathology emerges from:
-
Declining enzymatic degradation
-
Impaired glial phagocytosis
-
Reduced vascular efflux
-
Sleep-dependent glymphatic failure
-
Genetic modulation of lipid and immune pathways
This framework explains why anti-amyloid monoclonal antibodies show modest benefit only when administered very early, and why plaque removal alone does not restore cognition once synaptic loss and tau pathology dominate.
Clinical implications: moving beyond plaque removal
The future of amyloid-focused intervention lies in:
-
Early detection using plasma and CSF amyloid biomarkers
-
Preservation of clearance capacity, not just synthesis inhibition
-
Aggressive management of sleep and vascular risk
-
Integrated neurobiological approaches that protect synapses and networks
Amyloid remains central to Alzheimer’s disease—but only when understood as part of a dynamic clearance-failure model, not as a solitary toxic trigger.
About the Author
Dr. Srinivas Rajkumar T, MD (AIIMS, New Delhi), DNB, MBA (BITS Pilani)
Consultant Psychiatrist
Dr. Srinivas Rajkumar is a clinician-academic with a focused interest in amyloid biology, neurodegeneration, and early cognitive disorders. His clinical work integrates evidence-based psychiatry with contemporary neuroscience, emphasizing early identification of neurodegenerative risk, sleep-vascular-metabolic optimization, and brain-based interventions.
He practices at Mind & Memory Clinic, Apollo Clinic, Velachery (Chennai), where his work centers on memory complaints, mild cognitive impairment, dementia spectrum disorders, and neuropsychiatric syndromes, with a strong translational emphasis linking molecular mechanisms—such as amyloid metabolism—to real-world clinical care.
📍 Apollo Clinic, Velachery, Chennai
📞 +91-8595155808
✉️ srinivasaiims@gmail.com